
Work in science and science by itself are full of challenges. Recently most of these challenges were solved using different cross disciplinary approaches and in particular data science instruments. Additionally, contemporary biomedicine is strongly interconnected with programming and computational analysis, and therefore there are a few differences between ‘classic’ computer science and bioinformatics.
Together with new computational approaches, bioinformatics inherited new types of events, one of which is a hackathon. Now, what exactly is a hackathon? The main idea of a hackathon is an intense focused work on the challenges, which are needed to overcome. Also, events allow participants to share ideas and propose innovative solutions, improve their skill set and increase knowledge base.
Knowing about these benefits, the TranSYS ESRs were very excited about Copenhagen Bioinformatics Hackathon 2021 Protein Edition and had no hesitations to participate.
The focus of this hackathon was to build machine learning models to predict protein characteristics using structural or sequence data. The organisers announced 9 unique challenges related to protein biology and immunology, and approximately 200 participants in 60 teams from 26 different countries were doing their best to find the solutions.
Representing the TranSYS ITN, the three ambitious ESRs Giada Lalli (ESR 10), Maryna Korshevniuk (ESR12), and Sonja Katz (ESR 15) decided to unite their forces and enter the race as team ‘MadMaedchen’.
At the beginning it was quite difficult for us to decide what challenge to work on because almost half of them looked extremely inspiring and interesting. After long (but peaceful) discussions we finally agreed on a challenge to work on – it was Prediction of heavy and light chain antibody pairing, presented by: Oxford Protein Informatics Group (OPIG).
Choices are difficult – group brainstorming on which challenge to tackle for the Copenhagen Bioinformatics Hackathon 2021 Protein Edition
What was this challenge all about?
The binding site of antibodies is composed of two protein chains, known as the heavy and light chain. Not every combination of heavy and light chains forms a stable antibody, making the understanding and prediction of heavy and light chain pairing crucial in areas such as therapeutic antibody design. The focus of this challenge was to predict heavy and light chain pairings and to explore why certain pairings are possible and others not, using ~120.000 paired antibody sequences.[1]
A very simplistic baseline model was provided by the organisers and the task as hand was simple: improve this model in every imaginable way!
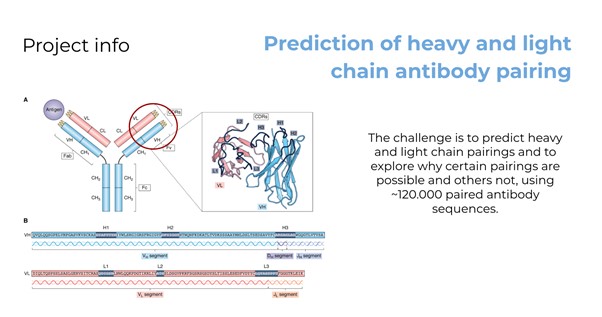 Overview of the challenge.
Overview of the challenge.
Our strategy
The MadMaedchen quickly detected some obvious weaknesses in the existing baseline model that, once addressed, could significantly improve the predictive power:
- Model selection and optimization
The baseline model featured a basic Random Forest classifier. By thorough hyperparameter tuning and classifier comparison using a cross-validation approach, we aimed on providing an unbiased estimate on the optimal machine learning model for this task
- Sequence embedding
Using knowledge from the field of natural language processing (NLP), sequence embedding models translating amino acid sequences into machine-readable format have been an intense focus of study. We sought to encode our data by using such a pre-trained embedding model (Yang et al., 2018), thereby reducing dimensionality but keeping the biological information contained in the sequence.
- Extending the feature space to biophysical properties
Machine learning tools allow integration of multiple different parameters, so we prepared a source for the next step in solving this challenge – we have analyzed more than 1000 biophysical properties of given amino acid sequences. We will use this as an auxiliary set of variables to further increase precision and stability of our prediction model.
Results & Conclusions
With only 36 hours dedicated to a challenge of this magnitude, we were able to partially realize some of the envisioned aspects, slightly improving the baseline model performance. However, the lack of computational power (and sleep 🙂 ) kept us from fully implementing all of our ideas. Despite not taking the crown, the #MadMaedchen held the record for several hours as the model with the best accuracy and AUC!
At the end of the day, all of us enjoyed having participated in this inspiring event, because what can be nicer than spending your weekend in front of the computer, frantically programming with your friends?
Our sincere thanks to the organising team, see you at the next hackathon! #BioHackathonDK2022
Data availability
The data set used in this challenge is publicly available here.
[1] Excerpt from: https://biohackathon.biolib.com/event/2021-protein-edition/#challenges